Xác định hồ sơ methyl hóa ADN bằng công nghệ giải trình tự thế hệ mới
Trong những năm gần đây, lĩnh vực di truyền học pháp y đang chứng kiến một sự chuyển dịch quan trọng từ phân tích ADN truyền thống sang nghiên cứu về di truyền ngoại gen – lĩnh vực nghiên cứu những thay đổi trong biểu hiện gen không làm biến đổi trình tự ADN. Trong số các cơ chế di truyền ngoại gen, methyl hóa ADN được cho là một công cụ tiềm năng mang tính cách mạng, mở ra những khả năng chưa từng có trong điều tra pháp y: ước lượng tuổi sinh học của người để lại dấu vết, phân biệt các cặp sinh đôi cùng trứng và xác định loại dịch cơ thể một cách chính xác.
Methyl hóa ADN là gì và tại sao nó quan trọng với pháp y?
Methyl hóa ADN là quá trình bổ sung một nhóm methyl vào vị trí số 5 của cytosine, thường xảy ra ở các dinucleotide CpG. Khoảng 60-90% các vị trí CpG trong hệ gen người được cho là ở trạng thái methyl hóa. Các vùng giàu CpG (được gọi là đảo CpG) thường liên quan đến vùng điều hòa của gen và đóng vai trò then chốt trong việc kiểm soát biểu hiện gen.
Điều làm nên giá trị pháp y của methyl hóa ADN nằm ở tính chất động của nó. Không giống như trình tự ADN cố định, mức độ methyl hóa có thể thay đổi theo thời gian dưới tác động của các yếu tố môi trường, lối sống và quá trình lão hóa sinh học. Chính đặc tính này biến methyl hóa ADN thành một kho thông tin vô giá, cho phép khai thác những dữ liệu mà phân tích ADN thông thường không thể cung cấp.
Ba ứng dụng chiến lược của phân tích methyl hóa ADN trong pháp y
Ước lượng tuổi sinh học
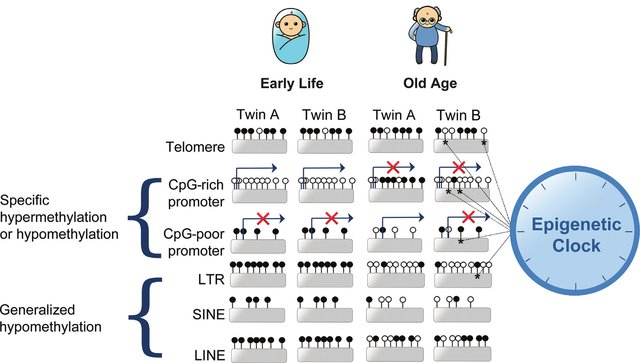
Khả năng ước lượng tuổi của người để lại dấu vết cung cấp thông tin tình báo quý giá cho cơ quan điều tra, đồng thời bổ sung thông tin cho các marker đặc điểm hình thái bên ngoài. Nhiều nghiên cứu trên toàn bộ hệ gen đã phát hiện các mô hình methyl hóa thay đổi theo tuổi tác. Đặc biệt, các marker liên quan đến gen ELOVL2 được nhiều nghiên cứu độc lập trên các quần thể khác nhau xác nhận có tương quan cao với tuổi sinh học.
Các mô hình dự đoán tuổi dựa trên một số ít marker CpG chủ yếu được phát triển cho mẫu máu với sai số khoảng ±5 năm. Tuy nhiên, các mô hình cho các dịch cơ thể khác như nước bọt hay tinh dịch vẫn còn hạn chế. Ngoài ra, sự khác biệt giới tính và sai số dự đoán cao hơn ở trẻ em, người già và những cá nhân có bệnh lý cũng là những thách thức cần tiếp tục nghiên cứu.

Phân biệt sinh đôi cùng trứng
Việc phân biệt các cặp sinh đôi cùng trứng trong giám định pháp y là bài toán nan giải vì chúng không thể phân biệt bằng kỹ thuật phân tích ADN chuẩn. Tuy nhiên, dù có sự giống nhau về mặt di truyền, các cặp sinh đôi cùng trứng thường biểu hiện những khác biệt về kiểu hình, có thể do ảnh hưởng của các yếu tố di truyền ngoại gen.
Nhiều nghiên cứu đã báo cáo sự khác biệt về mô hình methyl hóa ADN giữa các cặp sinh đôi cùng trứng. Sự khác biệt này càng rõ rệt ở những cặp sinh đôi lớn tuổi, sống xa nhau lâu hơn hoặc có tiền sử bệnh lý và sức khỏe khác biệt. Các nghiên cứu đã xác định được nhiều vùng và vị trí CpG tiềm năng cho mục đích phân biệt sinh đôi. Tuy nhiên, số lượng và mức độ khác biệt di truyền ngoại gen cần thiết để đạt được độ tin cậy thống kê vẫn chưa được xác định rõ và có thể phụ thuộc vào độ tuổi cũng như môi trường sống.
Xác định loại dịch cơ thể
Biến đổi di truyền ngoại gen đóng vai trò nền tảng trong điều hòa biệt hóa tế bào và biểu hiện gen, cho thấy tiềm năng xác định mô/dịch cơ thể dựa trên methyl hóa ADN. Năm 2011, Frumkin và cộng sự lần đầu tiên đề xuất sử dụng methyl hóa CpG khác biệt để xác định tinh dịch trong các hỗn hợp mẫu. Tiếp theo đó, nhiều nghiên cứu đã mô tả các marker di truyền ngoại gen và phương pháp phân tích cho các mô khác nhau bao gồm máu, nước bọt, tinh dịch, dịch âm đạo và máu kinh nguyệt.
Để có giá trị thực tiễn, các vị trí CpG ứng viên cần thể hiện sự biến động tối thiểu giữa các cá thể và trong cùng một cá thể đối với cùng một loại mô, đồng thời có mức methyl hóa khác biệt đáng kể giữa mô mục tiêu và tất cả các dịch cơ thể khác trong các điều kiện môi trường khác nhau.
Công nghệ phân tích methyl hóa ADN: Từ phương pháp truyền thống đến giải trình tự thế hệ mới
Các phương pháp phân tích methyl hóa ADN hiện nay có thể chia thành ba nhóm dựa trên cách xử lý ADN trước khi phân tích: sử dụng enzyme cắt nhạy cảm với methyl hóa, làm giàu bằng ái lực và biến đổi bằng bisulfite. Trong đó, phương pháp biến đổi bằng bisulfite được sử dụng phổ biến nhất trong nghiên cứu methyl hóa pháp y.
Biến đổi bisulfite là quá trình xử lý hóa học cytosine bằng natri bisulfite. Cytosine không methyl hóa được chuyển thành uracil thông qua quá trình khử amin, trong khi cytosine methyl hóa vẫn không thay đổi. Sau đó, ADN được khuếch đại bằng PCR, uracil được thay thế bằng thymine. Kết quả là các trình tự ADN khác nhau tùy thuộc vào trạng thái methyl hóa, cho phép xác định tỷ lệ methyl hóa tại từng vị trí CpG cụ thể.
Các phương pháp phân tích methyl hóa phổ biến hiện nay bao gồm pyrosequencing, kéo dài base đơn, phân tích đường cong nóng chảy độ phân giải cao và hệ thống EpiTYPER. Tuy nhiên, mỗi phương pháp đều có những hạn chế riêng về khả năng phân tích đa mồi, độ nhạy, thời gian và thiết bị yêu cầu.
Giải trình tự thế hệ mới: Lợi thế và thách thức
Công nghệ giải trình tự thế hệ mới là một giải pháp thay thế đầy triển vọng cho các phương pháp phân tích methyl hóa hiện tại. Với khả năng phân tích đồng thời hàng trăm mồi trong một phản ứng duy nhất, công nghệ này đặc biệt phù hợp với các mẫu pháp y có lượng ADN hạn chế.
Ưu điểm nổi bật
Khả năng đa mồi cho phép phát hiện đồng thời nhiều vị trí CpG từ các vị trí khác nhau trên hệ gen trong một phản ứng duy nhất – điều này vượt trội so với pyrosequencing vốn thường chỉ thực hiện được các phản ứng đơn lẻ. Hàng trăm sản phẩm PCR có thể được gộp chung để chuẩn bị thư viện và giải trình tự.
Độ nhạy phân tích cao hơn nhờ độ sâu đọc lớn, cho phép phát hiện mức methyl hóa nhỏ trong các mẫu không đồng nhất. Khả năng đọc trình tự dài hơn cũng mở ra triển vọng trong việc phân biệt sinh đôi cùng trứng, nơi các mô hình methyl hóa vùng có thể được quan tâm.
Đặc biệt, việc phát hiện methyl hóa ADN bằng giải trình tự thế hệ mới có tiềm năng kết hợp với các phân tích STR và SNP hiện có trong cùng quy trình chuẩn bị thư viện, tạo điều kiện tích hợp vào quy trình giám định pháp y hiện hành.
Thách thức cần vượt qua
Hạn chế kỹ thuật quan trọng nhất áp dụng cho mọi phương pháp dựa trên bisulfite là lượng ADN đầu vào lớn được khuyến nghị, khoảng 200-500 ng, do quá trình biến đổi bisulfite gây phân mảnh và thất thoát ADN đáng kể. Điều này đặc biệt khó khăn với mẫu pháp y vốn thường có lượng ADN rất nhỏ. Các nghiên cứu gần đây đã chỉ ra ảnh hưởng của hiệu ứng ngẫu nhiên lên kết quả methyl hóa từ mẫu có lượng ADN thấp, đòi hỏi sử dụng lặp lại và mô hình thống kê để diễn giải dữ liệu một cách tin cậy.
Biến đổi bisulfite cũng đặt ra thách thức cho khuếch đại PCR do độ phức tạp của ADN giảm, khiến mồi dễ bắt cặp chéo và tạo cấu trúc thứ cấp. Thiết kế mồi đóng vai trò then chốt với các nguyên tắc như tăng chiều dài mồi, sử dụng các mồi chứa cytosine không nằm trong CpG, tăng nhiệt độ bắt cặp và sử dụng polymerase hot start.
Về mặt xử lý dữ liệu, hiện chưa có phần mềm chuyên dụng hay quy trình xử lý chuẩn cho phân tích methyl hóa ADN dựa trên giải trình tự thế hệ mới, khác với các phương pháp SNaPshot, microarray hay pyrosequencing đã có quy trình ổn định. Việc lựa chọn công cụ tin sinh phù hợp, bao gồm căn chỉnh trình tự và xác định mức methyl hóa, hoàn toàn phụ thuộc vào người nghiên cứu, tiềm ẩn nguy cơ sai số nếu lựa chọn không phù hợp.
Kiểm soát sai số và nhiễu trong phân tích methyl hóa
Các nguồn sai số tiềm ẩn có thể xuất hiện trong suốt quy trình thực nghiệm và tin sinh, cần được quản lý chặt chẽ để tránh ảnh hưởng đến kết quả cuối cùng.
Hiệu quả chuyển đổi bisulfite: Quá trình chuyển đổi không hoàn toàn cytosine không methyl hóa dẫn đến ước tính quá cao mức methyl hóa. Việc sử dụng các mẫu chứng methyl hóa hoàn toàn và không methyl hóa là cách tốt nhất để đánh giá hiệu quả chuyển đổi.
Khuếch đại ưu tiên: PCR có thể ưu tiên khuếch đại một số đoạn ADN nhất định dựa trên trình tự và khả năng bắt cặp mồi. Điều này đặc biệt quan trọng vì cả marker methyl hóa và không methyl hóa cần được khuếch đại đồng đều để định lượng chính xác. Các vị trí CpG nên được tránh trong trình tự mồi nếu có thể.
Thuật toán căn chỉnh: Các thuật toán căn chỉnh có thể được chia thành hai nhóm: “có nhận biết methyl hóa” và “không nhận biết methyl hóa”. Nhóm có nhận biết thường căn chỉnh các trình tự methyl hóa hiệu quả hơn, dẫn đến ước tính quá cao mức methyl hóa. Nhóm không nhận biết được khuyến nghị sử dụng cho phân tích methyl hóa ADN pháp y.
Trùng lặp tín hiệu: Khi đọc cặp từ cả hai đầu của đoạn ADN chồng lên nhau, nếu không được xử lý đúng, vùng này sẽ được tính hai lần, làm sai lệch thông tin methyl hóa. Các công cụ tin sinh cần loại bỏ phần trình tự chồng lấn để tránh sai số.
Kết luận và triển vọng
Định lượng hồ sơ methyl hóa ADN có tiềm năng mang lại giá trị to lớn trong điều tra hình sự. Việc ứng dụng giải trình tự thế hệ mới trong phân tích methyl hóa ADN đích là một giải pháp thay thế khả thi cho các phương pháp hiện đang thống trị trong nghiên cứu pháp y. Cách tiếp cận này mang lại những lợi thế từ góc độ xét nghiệm pháp y nhờ khả năng phân tích đa mồi và tiềm năng kết hợp với các phân tích marker ADN khác.
Tuy nhiên, cần có những nỗ lực để thiết lập độ chính xác và khả năng tái lập của phương pháp này cho các mẫu pháp y. Các quy trình chuẩn mực tập trung vào giảm thiểu và hiệu chỉnh các sai số tiềm ẩn vốn có của biến đổi bisulfite và phân tích giải trình tự thế hệ mới cũng cần được phát triển trước khi các phương pháp này có thể được áp dụng thường quy trong giám định pháp y.
Dù vậy, công nghệ này đã cho thấy tiềm năng to lớn cho việc định lượng hồ sơ methyl hóa ADN pháp y trong tương lai, mở ra cánh cửa khai thác những lớp thông tin hoàn toàn mới từ các dấu vết sinh học, vượt xa giới hạn của phân tích ADN truyền thống.
Bài viết được tham khảo từ bài báo Evaluation of massively parallel sequencing for forensic DNA methylation profiling
Để tìm hiểu chi tiết thêm về ứng dụng và tính năng của các sản phẩm phục vụ cho lĩnh vực pháp y, xác định danh tính, huyết thống, vui lòng liên hệ chúng tôi để được hỗ trợ tư vấn trực tiếp.